Fleming Lab

The figure above illustrates the Gram-negative bacterial envelope. The outer membrane (OM) is composed of both phospholipid and lipopolysaccharide and is packed with β-barrel Outer Membrane Proteins. These OMP's are systhesized in the cytoplasm (bottom of figure) and transported across the inner membrane (IM) into the aqueous periplasm as unfolded outer membrane proteins (uOMP). The hydrophobic uOMP's form complexes with the chaperones Skp, SurA and FkpA which minimizes self-aggregation and allows diffusion of the uOMP's across the periplasm. Eventually, the uOMP's are handed off from the SurA-uOMP complex to BAM, a multi-chain protein that catalyzes the insertion and folding of uOMP's into the outer membrane. How these proteins cross the peptidyl glycan (PG) network is not known.
We are primarily an experimental lab that studies the intracellular transport and folding of bacterial outer membrane proteins describe above. We use computational methods to help us interpret and understand the experimental results.
OMPbioM
Outer membrane protein (OMP) biogenesis is critical to bacterial physiology because the cellular envelope is vital to bacterial pathogenesis and antibiotic resistance. This project integrates parameters and observations from both in vivo and in vitro experiments into a holistic computational model termed "Outer Membrane Protein Biogenesis Model" (OMPBioM).

Unfolded outer membrane protein (uOMP) structure
uOMPs must exist as an unfolded ensemble while interacting with a chaperone network in the periplasm of Gram-negative bacteria.
We use a combination of analytical ultracentrifugation and molecular simulations to build model ensembles of uOMPs that are defined by experimental properties. Early results suggest that uOMPs have differential intrinsic structure dependent on amino acid composition or sequence.
Ref: 1
Skp chaperone structure/function
Seventeen Kilodalton Protein (Skp) is a protein chaperone that facilitates the diffusion of uOMPs from the inner membrane to the outer membrane.
Skp is shaped like a jelly fish with three tentacles. Neutron scattering results show that this chaperone holds the unfolded uOMP in a cavity formed by the tentacles. The uOMP has the size and shape of a collapsed molten globule when bound to Skp.
Ref: 1
SurA chaperone structure/function
SurA is a protein chaperone that facilitates the diffusion of uOMPs from the inner membrane to the outer membrane. This chaperone has three domains on flexible linker regions and exists in multiple conformations. Two conformations are shown at left: open and collapsed.

Using neutron scattering, chemical cross linking, mass spectrometry, and computational modeling we discovered that when SurA binds to an uOMP it stabilizes an elongated form of the uOMP. Apparently this elongation is facilitated by the binding of multiple SurA on an uOMP as shown at left.
Ref: 1, 2, 3
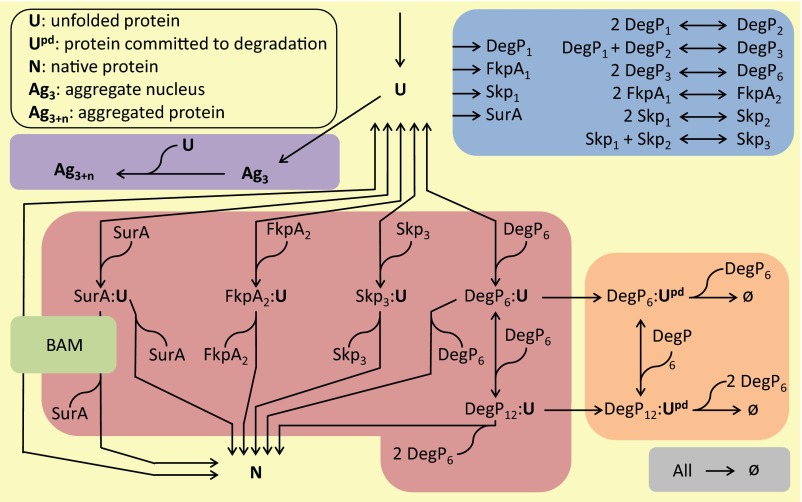
β-barrel structural properties
Transmembrane β-barrel proteins in the outer membrane of the bacterial envelope have high elastic stiffness and are laterally crowded. They are a major determinant of the properties of the bacterial envelope that resist mechanical stress.
Ref: 1, 2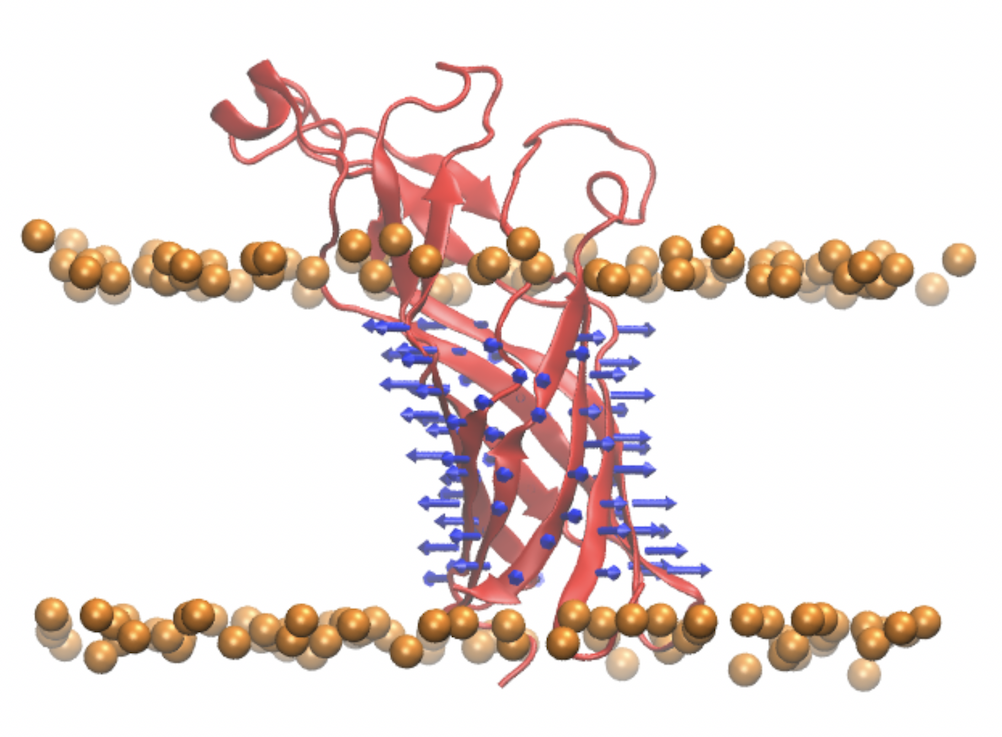
BAM
The β-Barrel Assembly Machine (BAM) is composed of five protein subunits; subunit A (surface rendering at left) is a transmembrane β-barrel itself with a long protein tail, and the other four subunits (ribbons rendering) are extrinisc proteins attached to the membrane by lipid tails.
The protein tail of BamA is flexible. It appears that the lipid-attached subunits affect the conformation of the BamA tail, an interaction that may increase the efficiency of the BAM catalytic action on OMP insertion and folding.
Ref: 1, 2
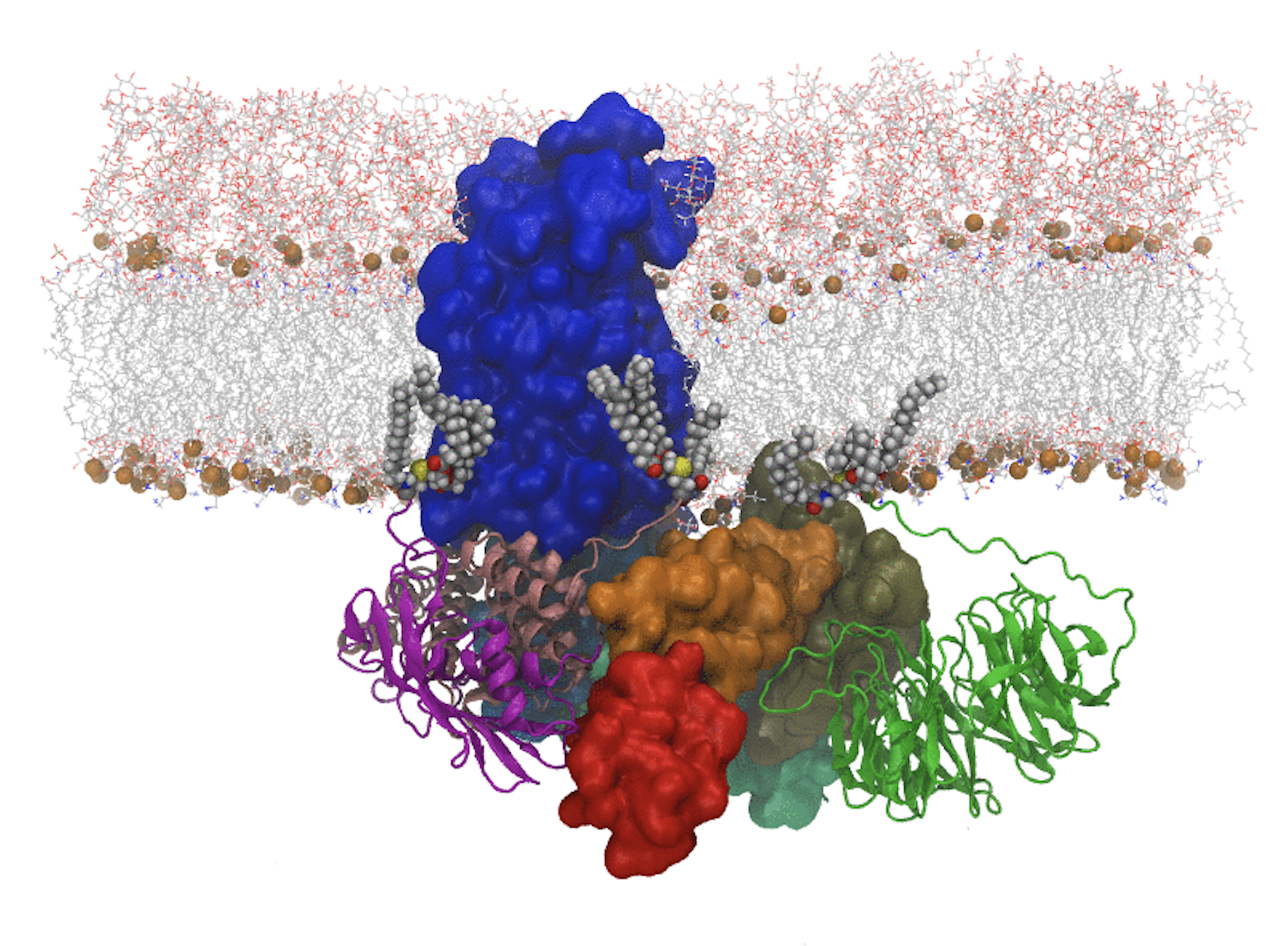
Biological amino acid hydrophobic scale
The process by which membrane proteins fold involves the burial of amino acid residue side chains into lipid bilayers. Both structure and function of membrane proteins depend on the magnitudes of side-chain transfer free energies. This project develops a water-to-bilayer transfer free energy scale (i.e., a "hydrophobicity scale") for the twenty natural amino acid side chains measured in the context of a native transmembrane protein and a phospholipid bilayer.
Ref: 1, 2, 3, 4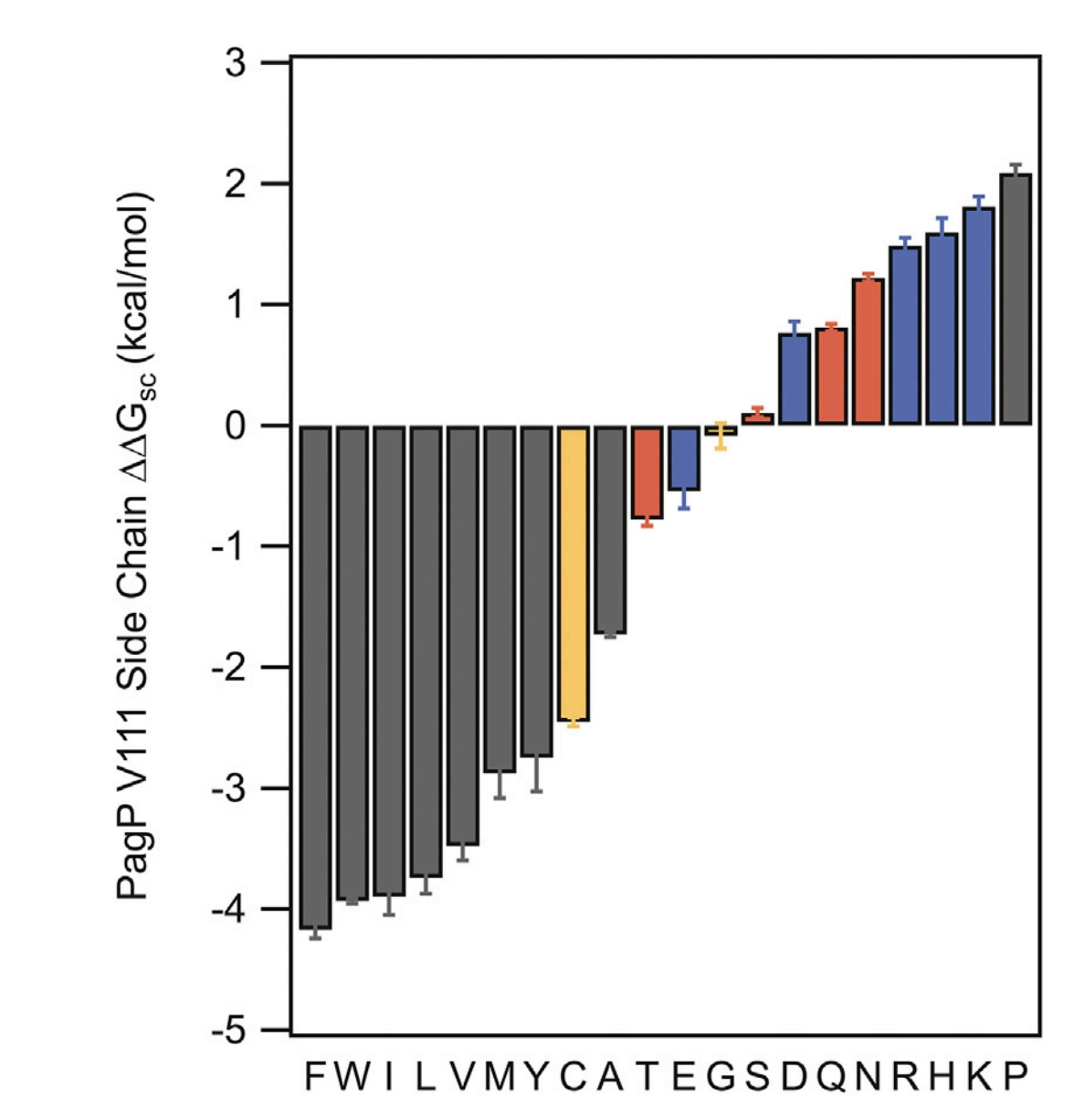
HullRad
HullRad is a computer program and webserver that calculates hydrodynamic properties of macromolecules from structure files.
It uses a convex hull representation of the molecule.
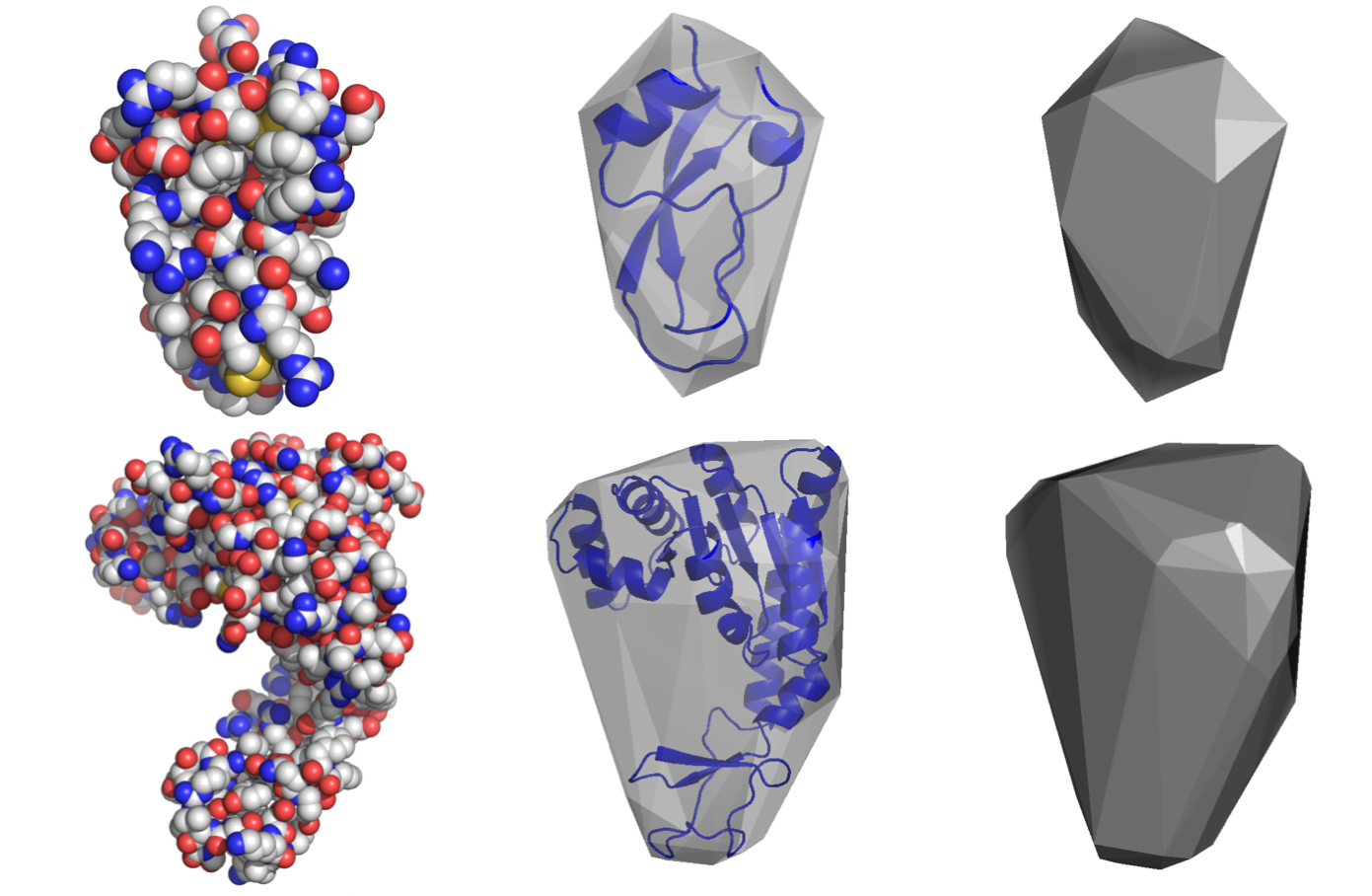
HullRad is so fast that one can calcuate the sedimentation coefficients for thousands of structures from molecular simulations in a very short time. This is useful for comparing to experimental hydrodynamic values of diffuse structural ensembles.
The figure at left shows a folded OMP (ribbon) in a detergent micelle (atomic spheres) with the time evolution of it's sedimentation coefficient below.
