 |
 |
 |
| Home | Research | People | Publications | Resources | PPG |
Mechanism of copper transport in human cells
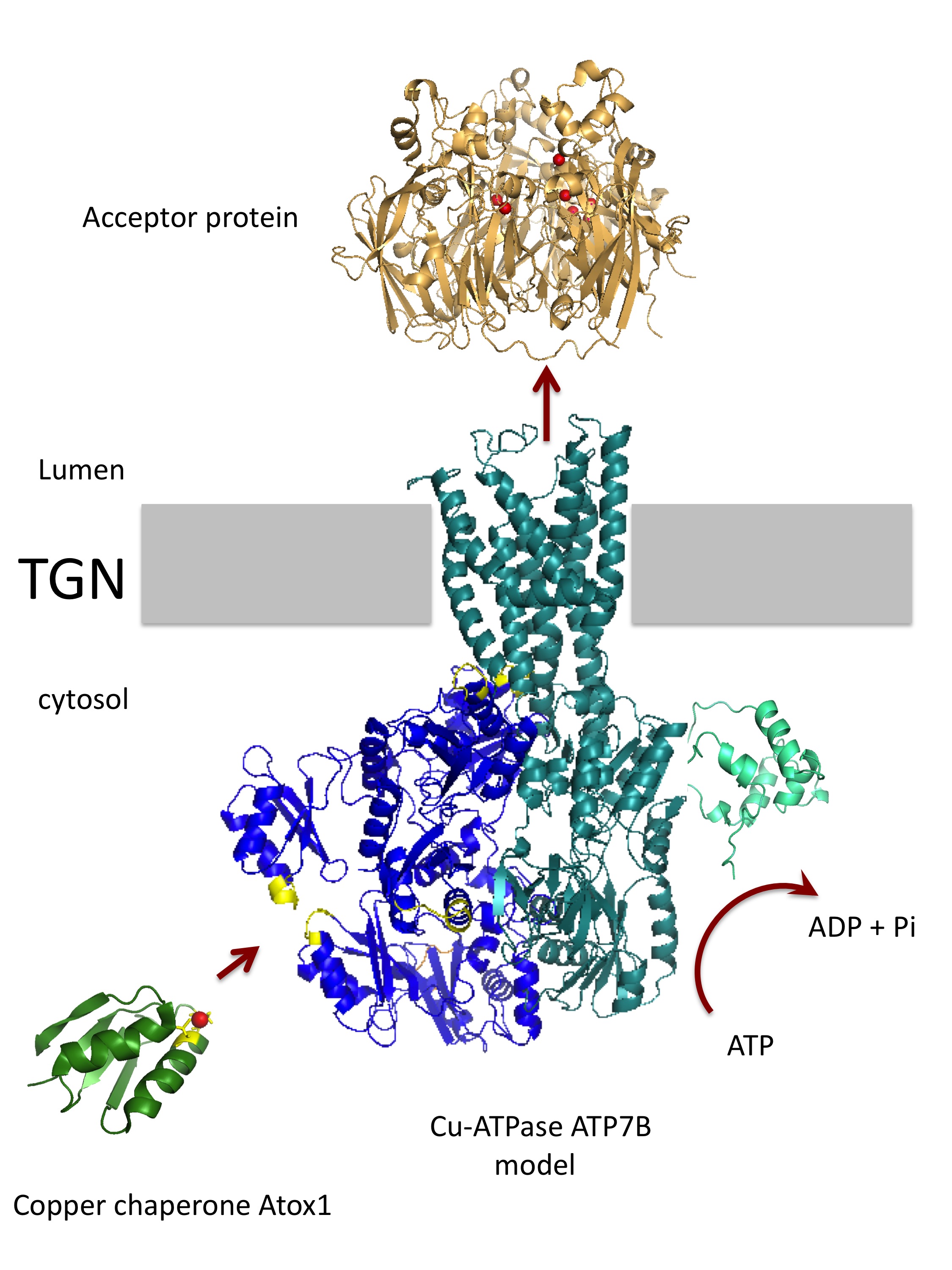 Copper is a redox active metal, which can readily donate and accept electrons. Copper-containing enzymes utilize
this property to perform reactions necessary for utilization of oxygen, detoxification of radicals, formation of the
tyrosyl quinone cofactor and other biochemical processes. For these reactions to take place in a cell, copper has
to be transported to the appropriate compartments (cytosol, secretory pathway, mitochondria) in which the copper-requiring
enzymes are located. This job is done by the copper transporters and small cytosolic proteins, copper chaperones, which
were named so for their ability to bind copper and thus guard cells from copper reactivity, while delivering it to
transporters and other acceptor proteins. In the laboratory, we study how the copper chaperone Atox1 transfers copper
to the transporters ATP7A and ATP7B located in the trans-Golgi network and in the vesicles of the endocytic pathway.
ATP7A and ATP7B are complex multi-domain proteins with at least 8 distinct copper binding sites.
We study how copper is transferred to these metal-binding sites, how various domains in ATP7A and ATP7B
communicate with each other to facilitate ion translocation across membranes and how copper is then incorporated
into the acceptor enzymes within the secretory pathway.
Further reading:
Copper is a redox active metal, which can readily donate and accept electrons. Copper-containing enzymes utilize
this property to perform reactions necessary for utilization of oxygen, detoxification of radicals, formation of the
tyrosyl quinone cofactor and other biochemical processes. For these reactions to take place in a cell, copper has
to be transported to the appropriate compartments (cytosol, secretory pathway, mitochondria) in which the copper-requiring
enzymes are located. This job is done by the copper transporters and small cytosolic proteins, copper chaperones, which
were named so for their ability to bind copper and thus guard cells from copper reactivity, while delivering it to
transporters and other acceptor proteins. In the laboratory, we study how the copper chaperone Atox1 transfers copper
to the transporters ATP7A and ATP7B located in the trans-Golgi network and in the vesicles of the endocytic pathway.
ATP7A and ATP7B are complex multi-domain proteins with at least 8 distinct copper binding sites.
We study how copper is transferred to these metal-binding sites, how various domains in ATP7A and ATP7B
communicate with each other to facilitate ion translocation across membranes and how copper is then incorporated
into the acceptor enzymes within the secretory pathway.
Further reading: The cross-talk between lipid and copper metabolisms mediated via regulation of RNA processing
 The cross-talk between lipid and copper metabolisms mediated via regulation of RNA processing.
Using Atp7b-/- mice, genomics and proteomics approaches, we discovered that lipid metabolism is especially
sensitive to copper elevation in the liver. We have also found that this sensitivity can be linked to
specific redistribution of copper to the nucleus and changes in the cellular RNA splicing machinery.
Our current goal is to understand the roles of several RNA processing proteins in copper metabolism and,
reciprocally, to dissect the mechanism through which copper triggers changes in the RNA splicing machinery.
Further reading:
The cross-talk between lipid and copper metabolisms mediated via regulation of RNA processing.
Using Atp7b-/- mice, genomics and proteomics approaches, we discovered that lipid metabolism is especially
sensitive to copper elevation in the liver. We have also found that this sensitivity can be linked to
specific redistribution of copper to the nucleus and changes in the cellular RNA splicing machinery.
Our current goal is to understand the roles of several RNA processing proteins in copper metabolism and,
reciprocally, to dissect the mechanism through which copper triggers changes in the RNA splicing machinery.
Further reading: Regulation of copper transport through hormonal signaling, intracellular trafficking,
and kinase-mediated phosphorylation
 Human copper metabolism is tightly coupled to other metabolic processes, as
well as various signaling events. Changes in the environment inside and outside
a cell require redistribution of copper between cellular compartments or export of
excess copper from cells. Human copper transporting ATPases ATP7A and ATP7B play the
key role in these processes. In response to various signals (such as changes in
copper concentration, hormonal signaling, or inflammation), these transporters
move from their basal location in trans-Golgi network to vesicles in order to
regulate copper delivery to metalloenzymes as well as facilitate copper efflux.
We discovered that the intracellular trafficking of ATP7B was coupled to changes
in phosphorylation of this transporter by kinases, and we are now investigating
the role of phosphorylation in targeting of ATP7B to distinct cellular compartments.
We have also discovered a copper-independent trafficking of ATP7A in adipocytes, and
we are interested in the role of this process in physiology of adipose tissue. Recently,
using siRNA screening strategy, we identified a set of new regulators for copper-transporting
ATPase ATP7A. By characterizing these proteins we hope to build a comprehensive regulatory network for ATP7A.
Further reading:
Human copper metabolism is tightly coupled to other metabolic processes, as
well as various signaling events. Changes in the environment inside and outside
a cell require redistribution of copper between cellular compartments or export of
excess copper from cells. Human copper transporting ATPases ATP7A and ATP7B play the
key role in these processes. In response to various signals (such as changes in
copper concentration, hormonal signaling, or inflammation), these transporters
move from their basal location in trans-Golgi network to vesicles in order to
regulate copper delivery to metalloenzymes as well as facilitate copper efflux.
We discovered that the intracellular trafficking of ATP7B was coupled to changes
in phosphorylation of this transporter by kinases, and we are now investigating
the role of phosphorylation in targeting of ATP7B to distinct cellular compartments.
We have also discovered a copper-independent trafficking of ATP7A in adipocytes, and
we are interested in the role of this process in physiology of adipose tissue. Recently,
using siRNA screening strategy, we identified a set of new regulators for copper-transporting
ATPase ATP7A. By characterizing these proteins we hope to build a comprehensive regulatory network for ATP7A.
Further reading: Metallochaperone Atox1, copper transfer mechanism
Understanding Wilson’s disease pathology
 Wilson's disease is caused by mutations in copper-transporting ATPase
ATP7B. Elevated copper gradually induces a large spectrum of severe abnormalities,
including liver fibrosis, neuronal degeneration, and behavioral changes.
At present, the molecular events that accompany copper accumulation in
tissues are poorly understood. The goal of our studies is to dissect
the biochemical basis of pathological changes associated with abnormal
accumulation of
copper
in human cells. To understand these events we are currently identifying
the major targets of inborn copper toxicity using the recently developed
ATP7B knock-out
mouse (an animal model for Wilson's disease (Buiakova et
al., 1999) [PDF]), oligonucleotide
microarray technology and real-time PCR.
Wilson's disease is caused by mutations in copper-transporting ATPase
ATP7B. Elevated copper gradually induces a large spectrum of severe abnormalities,
including liver fibrosis, neuronal degeneration, and behavioral changes.
At present, the molecular events that accompany copper accumulation in
tissues are poorly understood. The goal of our studies is to dissect
the biochemical basis of pathological changes associated with abnormal
accumulation of
copper
in human cells. To understand these events we are currently identifying
the major targets of inborn copper toxicity using the recently developed
ATP7B knock-out
mouse (an animal model for Wilson's disease (Buiakova et
al., 1999) [PDF]), oligonucleotide
microarray technology and real-time PCR.
Copper homeostasis in the brain (Coming soon)
Department of Physiology
Johns Hopkins University
725 N. Wolfe Street
203 Hunterian
Baltimore, MD 21205
(410) 614-4661 (tel)
(410) 955-0461 (fax)